
How Quantitative is Traditional ChIP-Seq?
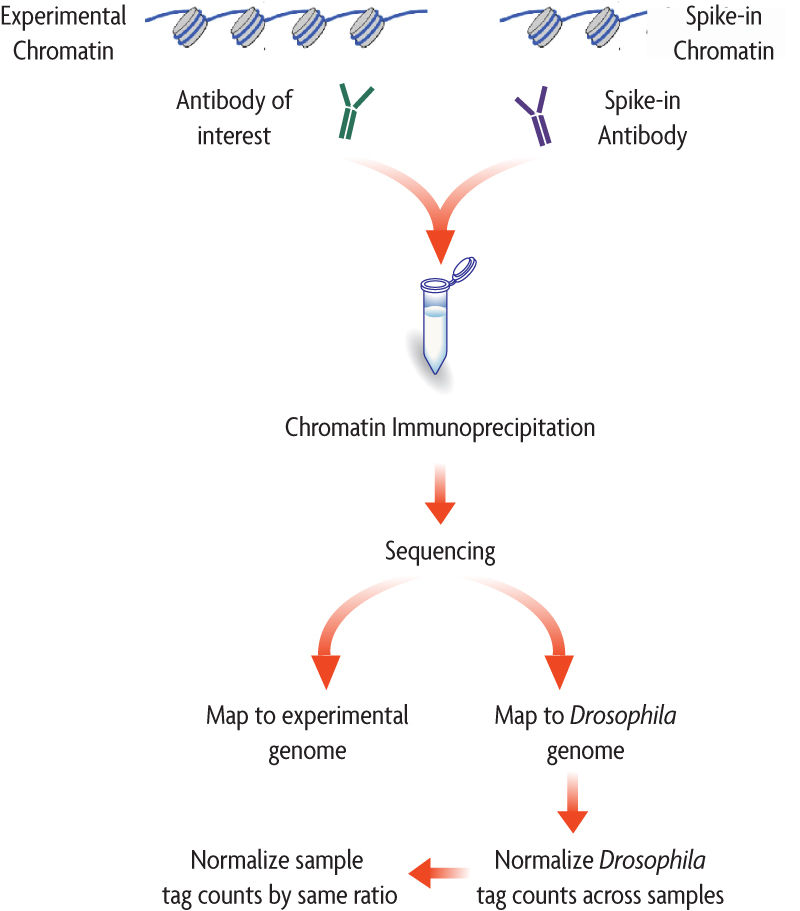
ChIP-Seq is a powerful tool for genome-wide identification of transcription factor binding sites and histone post-translational modifications, but its inherent semi-quantitative nature can potentially limit researchers’ ability to compare differences between samples in some cases (described in greater detail in this webinar). However, by “spiking in” a small amount of exogenous chromatin into your samples prior to the ChIP reaction, you can normalize the signal from your experimental samples to this control in your final sequencing data. This approach allows researchers to be more confident than ever in the accuracy of their inter-sample comparisons.
How Can Spike-In Normalization Improve ChIP-Seq Analysis?
One particularly compelling example of the importance of ChIP-Seq normalization is illustrated in experiments investigating the genome-wide localization of the repressive histone mark H3K27me3 in the presence of epigenetic inhibitors. H3K27me3 has one identified methyltransferase (EZH2) responsible for catalyzing methyl group addition. However, EZH2 inhibition did not appear to significantly affect H3K27me3 levels in a ChIP-Seq experiment (top two tracks in Figure 1).
Following normalization to spike-in, the significant loss of H3K27me3 becomes apparent (bottom two tracks in Figure 1). Therefore, spike-in can reveal changes in histone modifications that would otherwise be masked by experimental artifacts.
